Приём пищи человеком происходит иногда со значительными интервалами, поэтому в организме выработались механизмы депонирования энергии. ТАГ (нейтральные жиры) – наиболее выгодная и основная форма депонирования энергии. Депонированный жир может обеспечивать организм энергией при голодании в течение длительного времени (до 7-8 недель). Синтез ТАГ происходит в абсорбтивный период в печени и жировой ткани. Но если жировая ткань – только место депонирования жира, то печень выполняет важную роль превращения части углеводов, поступающих с пищей, в жиры, которые затем секретируются в кровь в составе ЛПОНП и доставляются в другие ткани. Непосредственными субстратами в синтезе жиров являются ацил-КоА и глицерол-3-фосфат. Метаболический путь синтеза жиров в печени и жировой ткани одинаков, за исключением разных путей образования глицерол-3-фосфата.
Печень – основной орган, где идет синтез жирных кислот из продуктов гликолиза. В гладком эндоплазматическом ретикулюме гепатоцитов жирные кислоты активируются и сразу же используются для синтеза ТАГ, взаимодействуя с глицерол-3-фосфатом. Синтезированные жиры упаковываются в ЛПОНП и секретируются в кровь.
В жировой ткани для синтеза ТАГ используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП. Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом. Кроме жирных кислот, поступающих в адипоциты из крови, в этих клетках идет и синтез жирных кислот из продуктов распада глюкозы. Молекулы ТАГ в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул.
Регуляция синтеза триацилглицеролов
В абсорбтивный период при увеличении соотношения инсулин/глюкагон активируется синтез ТАГ в печени. В жировой ткани индуцируется синтез липопротенлипазы (ЛПЛ), т.е в этот период активируется поступление жирных кислот в адипоциты. Одновременно инсулин активирует белки-переносчики глюкозы – ГЛЮТ-4, что ведет к увеличению поступления глюкозы в адипоциты и активации там гликолиза. В результате образуются необходимые для синтеза жиров глицерол-3-фосфат и активированные жирные кислоты. В печени в результате действия инсулина увеличивается количество и активность регуляторных ферментов гликолиза, пируватдегидрогеназного комплекса, а также ферментов, участвующих в синтезе жирных кислот из ацетил-КоА. Итогом этих изменений является увеличение синтеза ТАГ и секреция их в кровь в составе ЛПОНП. ЛПОНП доставляют жиры в капилляры жировой ткани, где действие ЛПЛ обеспечивает быстрое поступление жирных кислот в адипоциты, где они депонируются в составе ТАГ.
Мобилизация жиров, т.е. гидролиз до глицерола и жирных кислот, происходит в постабсорбтивный период, при голодании и активной физической работе. Процесс осуществляется под действием гормончувствительной ТАГ-липазы. Этот фермент отщепляет одну жирную кислоту у первого углеродного атома глицерола с образованием диацилглицерола, а затем другие липазы гидролизуют его до глицерола и жирных кислот, которые поступают в кровь. Глицерол как водорастворимое вещество транспортируется кровью в свободном виде, а жирные кислоты – в комплексе с белком плазмы альбумином.
Регуляция мобилизации триацилглицеролов
Мобилизация депонированных ТАГ стимулируется глюкагоном и адреналином, и, но в гораздо меньшей степени, соматотропным гормоном и кортизолом. В постабсорбтивный период и при голодании глюкагон, действуя на адипоциты через аденилатциклазную систему, активирует гормончувствительную липазу, что инициирует липолиз и выделение жирных кислот и глицерола в кровь. При физической активности увеличивается секреция адреналина, который также через аденилатциклазную систему активирует липолиз. В настоящее время предполагается, что действие адреналина двояко: при низких концентрациях в крови преобладает его антилиполитическое действие через α2-рецепторы, а при высокой – преобладает липолитическое действие через β-рецепторы.
В результате мобилизации ТАГ концентрация жирных кислот в крови увеличивается приблизительно в 2 раза, но они достаточно быстро утилизируются. Для мышц, сердца, почек, печени при голодании или физической работе жирные кислоты становятся важным источником энергии. Печень перерабатывает часть жирных кислот в кетоновые тела, используемые мозгом, нервной и некоторыми другими тканями как источники энергии. Когда постабсорбтивный период сменяется абсорбтивным, инсулин через промежуточные механизмы подавляет активность гормончувствительной липазы и распад жиров останавливается.
Ожирение
Состояние, когда масса тела на 20% превышает идеальную для данного индивидуума, считают ожирением. Оно развивается, когда в жировой ткани преобладают процессы липогенеза. Образование адипоцитов происходит во внутриутробном состоянии, начиная с последнего триместра беременности, и заканчивается в препубертатный период. После этого жировые клетки могут увеличиваться в размерах при ожирении или уменьшаться при похудании, но их количество не изменяется в течение жизни. Одна из классификаций ожирения основана на размерах и количестве адипоцитов. При повышении общего числа этих клеток говорят о гиперпластическом ожирении (развивающемся в младенческом возрасте, наследственном); увеличесние размеров адипоцитов ведет гипертрофическому ожирению. Согласно другой классификации, выделяют первичное и вторичное ожирение.
Первичное ожирение развивается в результате алиментарного дисбаланса – избыточной калорийности питания по сравнению с расходами энергии. Причинами этого в 80% случаев являются генетические нарушения, далее в списке причин следуют состав и количество потребляемой пищи, уровень физической активности и психологические факторы. Метаболические различия между тучными и худыми людьми до настоящего времени не могут быть определены однозначно. Среди причин, объясняющих эти различия, называют то, что у людей, склонных к ожирению, более эффективный метаболизм, разное соотношение аэробного и анаэробного гликолиза, различия в активности Na+/K+-АТФазы. Установлено, что у человека и животных есть ген ожирения – obese gene. Продуктом экспрессии этого гена является белок лептин, который синтезируется и секретируется адипоцитами и взаимодействует с рецепторами гипоталамуса. В результате его действия снижается секреция нейропептида Y, стимулирующего потребление пищи. У большей части больных ожирением имеется генетический дефект рецепторов лептина в гипоталамусе, у некоторых дефектен сам ген. Но в итоге секреция нейропептида Y продолжается, что ведет к увеличению аппетита и, соответственно, к увеличению массы тела.
Вторичное ожирение – ожирение, развивающееся в результате какого-либо заболевания, чаще всего эндокринного. Например, к развитию ожирения приводят гипотиреоз, синдром Иценко-Кушинга и гипогонадизм.
Обмен жирных кислот
Высвобождающиеся при липолизе жирные кислоты поступают в кровоток и транспортируются в связанном с сывороточными альбуминами состоянии. Поступление СЖК сопровождается появлением в плазме также и глицерола. Глицерол может участвовать в глюконеогенезе или включаться в гликолитический путь с предварительным образованием глицерол-3-фосфата.
После того, как жирные кислоты поступают в клетку, они активируются путем образования кофермент А-производных:
RCOOH + HSKoA + ATФ ® RCO ~КоА + АМФ +ФФН
Реакцию катализируют ферменты ацил-КоА-синтетазы. Они находятся как в цитозоле, так и в матриксе митохондрий и отличаются по специфичности к жирным кислотам с различной длиной углеводородной цепи. Жирные кислоты с длиной цепи от 2 до 4 атомов углерода могут проникать в матрикс митохондрий путем диффузии. Активация таких кислот происходит в матриксе митохондрий. Жирные кислоты с длинной цепью, которые преобладают в организме человека, активируются ацил-КоА-синтетазами, расположенными на внешней мембране митохондрий.
β-Окисление жирных кислот происходит в матриксе митохондрий, поэтому после активации эти субстраты должны транспортироваться внутрь митохондрий. Этот процесс осуществляется с помощью карнитина, который поступает с пищей или синтезируется из незаменимых аминокислот лизина и метионина.
В наружной мембране митохондрий (Рис. 20.1) находится фермент карнитинацилтрансфераза I, катализирующий реакцию с образованием ацилкарнитина. Образовавшийся ацил-карнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется с помощью карнитинацилкарнитин-транслоказы на внутреннюю поверхность внутренней мембраны митохондрий, где фермент карнитинацилтрансфераза II катализирует перенос ацила на внутримитохондриальный КоА. После этого ацил-КоА включается в реакции β-окисления. Свободный карнитин возвращается в межмембранное пространство той же транслоказой.
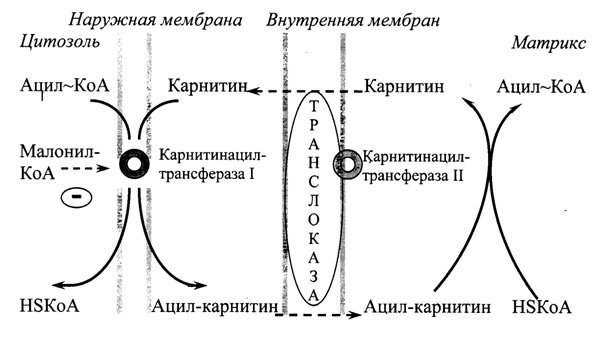
Рис. 20.1. Перенос длинноцепочечных жирных кислот через мембраны митохондрий.
β-Окисление жирных кислот – специфический путь катаболизма жирных кислот, протекающий в матриксе митохондрий только в аэробных условиях и заканчивающийся образованием ацетил-КоА. Водород из реакций β-окисления поступает в ЦТД, а ацетил-КоА окисляется в цикле трикарбоновых кислот, также поставляющем водород для ЦТД. Поэтому β-окисление жирных кислот является важнейшим метаболическим путем, обеспечивающим синтез АТФ в дыхательной цепи.
Продуктами каждого цикла β-окисления являются ФАДН2, НАДН и ацетил-КоА. Остаток кислоты, который входит в каждый последующий цикл, короче на 2 углеродных атома. В последнем цикле, когда остаётся жирная кислота из 4 атомов углерода, образуются сразу 2 молекулы ацетил-КоА. Суммарное уравнение β-окисления пальмитоил-КоА может быть представлено так:
С15Н31СО-КоА +7 ФАД+ 7 НАД++7 HSКоА ®8 СН3СО-КоА+7 ФАДН2+7 (НАДН+Н+).
Энергетический выход в этом случае составляет 131 молекулу АТФ (21 АТФ образуется при окислении каждой из 7 молекул НАДН в ЦТД, 14 – при окислении каждой из 7 молекул ФАДН2 в ЦТД, синтез 96 молекул АТФ обеспечивается окислением 8 молекул ацетил-КоА в ЦТК). С учетом расхода 1 молекулы АТФ на активцию кислоты, чистый энергетический выход окисления пальмитата составляет 130 АТФ. Окисление жирных кислот – важный источник энергии в тканях с высокой активностью ЦТК и дыхательной цепи (скелетные и сердечная мышцы, почки). Эритроциты, в которых отсутствуют митохондрии, не могут окислять жирные кислоты. Эти соединения не служат источником энергии для головного мозга, так как жирные кислоты не проходят через гематоэнцефалический барьер.
Регуляция скорости β-окисления. Скорость процесса регулируется потребностью клетки в энергии (соотношениями АТФ/АДФ, НАДН/НАД+). Скорость β-окисления зависит и от доступности субстрата, т.е. от количества жирных кислот, поступающих в митохондрии. Концентрация СЖК в крови повышается при активации липолиза. В этих условиях жирные кислоты становятся преимущественным источником энергии для мышц и печени, так как в результате β-окисления образуются НАДН и ацетил-КоА, ингибирующие пируватдегидрогеназный комплекс. Таким образом, использование жирных кислот как основного источника энергии в мышечной ткани и печени сберегает глюкозу для нервной ткани и эритроцитов.
Скорость β-окисления зависит также от активности карнитинацилтрансферазы I. В печени этот фермент ингибируется малонил-КоА, образующимся при биосинтезе жирных кислот. То есть малонил-КоА ингибирует деградацию жирных кислот, чем способствует их использованию для синтеза жира.
Другие типы окисления жирных кислот. β-Окисление является основным путем катаболизма жирных кислот, но помимо него встречаются α-окисление и ω–окисление. α-Окисление представляет собой последовательное отщепление одноуглеродных фрагментов, выделяющихся в виде СО2 от карбоксильного конца молекулы. Такому типу окисления подвергаются жирные кислоты с цепью более 20 углеродных атомов (характерны для липидов нервной ткани), а также жирные кислоты с разветвленной углеродной цепью (поступают с пищей). ω–Окисление жирных кислот в норме весьма незначительно, происходит оно в микросомах печени. Первоначальная стадия катализируется монооксигеназой, которая тебует наличия НАДФН, О2 и цитохрома Р-450. Группа -СН3 при этом превращается в -СН2ОН, затем окисляется до –СООН. Образовавшаяся дикарбоновая кислота может быть укорочена с любого конца путем реакций β-окисления.
Окисление ненасыщенных жирных кислот идет обычным путем, до тех пор, пока двойная связь не окажется между третьим и четвертым атомами углерода. После этого фермент еноил-КоА-изомераза перемещает двойную связь из положения 3-4 в положение 2-3 и изменяет цис-конформацию двойной связи на транс-, которая требуется для β-окисления. В этом цикле β-окисления первая реакция дегидрирования не происходит, так как двойная связь в радикале жирной кислоты уже имеется. Далее циклы β-окисления продолжаются, не отличаясь от обычного пути.
Жирные кислоты с нечетным числом углеродных атомов на конечном этапе β-окисления образуют ацетил-КоА и пропионил-КоА. Трехуглеродный фрагмент в ходе трех реакций превращается в сукцинил-КоА – метаболит ЦТК.
Ацетил-КоА, образующийся при β-окислении жирных кислот, расщеплении кетогенных аминокислот и окислительном декарбоксилировании пирувата служит исходным субстратом для ряда важнейших метаболических путей: 1) окисление в ЦТК, 2) образование кетоновых тел, 3) биосинтез холестерола, 4) биосинтез жирных кислот.
Обмен кетоновых тел
При голодании, длительной физической нагрузке и в случаях, когда клетки не получают достаточного количества глюкозы (желудочно-кишечные расстройства у детей, диета с низким содержанием углеводов, почечная глюкозурия, сахарный диабет), в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, увеличивается скорость b-окисления. Активность ЦТК в этих условиях снижена, так как ЩУК используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза ацетоацетата. Это вещество может выделяться в кровь или превращаться в печени в другое кетоновое тело – b-гидроксибутират путем восстановления. В клетках печени при активном b-окислении создается высокая концентрация НАДН. Это способствует превращению большей части ацетоацетата в b-гидроксибутират, поэтому основное кетоновое тело крови – именно b-гидроксибутират. При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путем организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, и вызывают ацидоз, так как являются кислотами. Скорость синтеза кетоновых тел зависит от активности 3-гидрокси-3-метилглутарил-КоА-синтазы (ГМГ-КоА-синтазы). Это индуцируемый фермент, его синтез увеличивается при повышении концентрации жирных кислот в крови. ГМГ-КоА-синтаза ингибируется высокими концентрациями свободного КоА. В норме образуется небольшое количество кетоновых тел (их содержание в крови составляет 10-30 мг/л, т.е. до 0,2 ммоль/л). В печени ацетоацетат не может окисляться, поэтому с током крови он попадает в скелетные мышцы, сердце, мозг, которые способны превращать ацетоуксусную кислоту вновь в ацетил-КоА.
Содержание кетоновых тел в крови увеличивается тогда, когда основным источником энергии для организма служат жирные кислоты – при длительной мышечной работе, голодании, сахарном диабете.

Рис. 20.2. Образование, утилизация и выведение кетоновых тел (главный путь показан непрерывными стрелками)
Увеличение концентрации кетоновых тел в крови называют кетонемией, выделение кетоновых тел с мочой – кетонурией. Накопление кетоновых тел в организме приводит в кетоацидозу: уменьшению щелочного резерва, а в тяжелых случаях – к сдвигу рН, так как b-гидроксибутират и ацетоацетат являются водорастворимыми органическими кислотами, способными к диссоциации. Ацидоз достигает опасных величин при сахарном диабете. Содержание кетоновых тел в крови при этом заболевании увеличивается в 100 и более раз, достигая концентрации 4-5 г/л. Тяжелая форма ацидоза – одна из основных причин смерти при сахарном диабете.
Синтез жирных кислот
Синтез жирных кислот происходит в основном в печени, в меньшей степени – в жировой ткани и лактирующей молочной железе. Гликолиз и последующее окислительное декарбоксилирование пирувата способствуют увеличению концентрации ацетил-КоА в матриксе митохондрий. Синтез же жирных кислот происходит в цитозоле, куда и должен быть транспортирован субстрат. Для этого в матриксе митохондрий ацетил-КоА конденсируется со ЩУК с образованием цитрата. Затем транслоказа переносит цитрат в цитоплазму. Это происходит только при увеличении количества цитрата в митохондриях, когда изоцитратдегидрогеназа и a-кетоглутаратдегидрогеназа ингибированы высокими концентрациями НАДН и АТФ. Такая ситуация создается в абсорбтивном периоде, когда клетка печени получает достаточное количество источников энергии. В цитоплазме цитрат расщепляется до ЩУК и ацетил-КоА. Последний служит исходным субстратом для синтеза жирных кислот, а ЩУК под действием малатдегидрогеназы превращается в малат, который при участии малик-фермента образует пируват. Пируват транспортируется обратно в матрикс митохондрий.
Первая реакция синтеза жирных кислот – превращение ацетил-КоА в малонил-КоА, осуществляемое ацетил-КоА-карбоксилазой, определяет скорость всех последующих реакций синтеза жирных кислот.
Далее синтез жирных кислот продолжается на мультиферментном комплексе – синтазе жирных кислот. Этот фермент состоит из 2 идентичных протомеров, каждый из которых имеет доменное строение и, соответственно, 7 центров, обладающих разными каталитическими активностями (ацетилтрансацилаза, малонилтрансацилаза кетоацилсинтаза, кетоацилредуктаза, гидратаза, еноил-редуктаза, тиоэстераза) и ацилпереносящий белок (АПБ). АПБ не является ферментом, его функция связана только с переносом ацильных радикалов. В процессе синтеза важную роль играют SH-группы. Одна из них принадлежит 4-фосфопантетеину, входящему в состав АПБ, вторая – цистеину кетоацилсинтазы. Протомеры синтазы жирных кислот расположены «голова к хвосту». Несмотря на то, что каждый мономер содержит все активные центры, функционально активен комплекс из двух протомеров. Поэтому реально синтезируются одновременно 2 жирных кислоты (в схемах для упрощения изображают синтез только одной молекулы).
Этот комплекс последовательно удлиняет радикал жирной кислоты на 2 углеродных атома, донором которых служит малонил-КоА. Циклы реакций повторяются до тех пор, пока не образуется радикал пальмитиновой кислоты, который под действием тиоэстеразного центра гидролитически отделяется от ферментного комплекса, превращаясь в свободную пальмитиновую кислоту. В каждом цикле биосинтеза пальмитиновой кислоты проходят 2 реакции восстановления, донором водорода в которых служит НАДФН.
Регуляция синтеза жирных кислот. Регуляторный фермент синтеза жирных кислот – ацетил-КоА-карбоксилаза. Его активность регулируется двумя способами.
1. Ассоциация/диссоциация комплексов субъединиц. В неактивной форме ацетил-КоА-карбоксилаза представляет собой отдельные комплексы, каждый из которых состоит из 4 субъединиц. Активатор фермента – цитрат – стимулирует объединение комплексов, ингибитор – пальмитоил-КоА – вызывает их диссоциацию.
2. Фосфорилирование/дефосфорилирование ацетил-КоА-карбоксилазы. В постабсорбтивном состоянии или при физической работе глюкагон или адреналин через аденилатциклазную систему активируют протеинкиназу А и стимулируют фосфорилирование субъединиц ацетил-КоА-карбоксилазы. Фосфорилированный фермент неактивен, синтез жирных кислот останавливается. В абсорбтивный период инсулин активирует фосфатазу, и ацетил-КоА-карбоксилаза переходит в дефосфорилированное состояние. Затем под действием цитрата происходит полимеризация протомеров фермента, и он становится активным.
Ещё одним способом усиления синтеза жирных кислот является индукция синтеза ферментов этого метаболического пути. Такое происходит при длительном потреблении богатой углеводами и бедной жирами пищи, когда инсулин стимулирует индукцию синтеза ацетил-КоА-карбоксилазы, синтазы жирных кислот, цитратлиазы и изоцитратдегидрогеназы.
Из пальмитиновой кислоты могут синтезироваться более длинные, а также ненасыщенные жирные кислоты. Удлинение пальмитиновой кислоты может происходить:
а) в митохондриях за счет присоединения ацетил-КоА по пути, обратному b-окислению, с использованием НАДФН, а не ФАДН2;
б) в микросомах за счет малонил-КоА и НАДФН. Процесс напоминает функционирование синтазного комплекса в цитозоле, только промежуточные продукты не связываются с АПБ.
Введение двойных связей в структуру жирных кислот происходит также в микросомах с помощью оксидаз, при этом используются НАДФН и О2.
Поможем написать любую работу на аналогичную тему