Высокая скорость и эффективность ферментативных реакций является результатом значительного снижения энергии активации. Снижение энергии активации и увеличение скорости реакции биокатализаторами достигается во-первых тем, что многие ферменты сорбируют субстраты на своей поверхности, затем строго ориентированно " подают" их на активный центр, где субстрат присоединяется (в зонах связывания) ионными, водородными связями и силами гидрофобного взаимодействия. Причем, чем выше специфичность фермента, тем больше точек связывания.
Во-вторых, многоточечное связывание субстрата (субстратов) используется ферментом на сближение и удержание в контакте реагирующих групп субстрата (субстратов) и каталитического участка столько времени, сколько необходимо для превращения субстрата (субстратов) в продукт (продукты).
В третьих, увеличению скорости реакции способствуют конформационные изменения белковой части ES , вызывающие напряжение, деформацию, дестабилизацию разрываемой связи в субстрате.
В четвертых, дестабилизация разрываемой связи наступает при переносе заряженного субстрата из окружающего водного раствора в более гидрофобную среду активного центра фермента.
В пятых, многие ферменты осуществляют полифункциональный катализ. В их активном центре, на связь субстрата одновременно или последовательно воздействуют несколько разнообразных R-групп остатков аминокислот белковой части и групп кофактора, в результате происходит поляризация а затем и разрыв атакуемой связи.
В шестых, многие группы в активных центрах ферментов функционируют как кислоты (доноры Н+) или основания (акцепторы Н+). Особенно эффективен обобщенный кислотно-основной катализ. Он дает увеличение скорости в 10-100 раз. В качестве кислотно-основных катализаторов функционируют радикалы глутаминовой, аспаргиновой, гистидина, лизина, тирозина и др. остатков аминокислот. В протонированной форме они являются кислотными, в непротонированной - основными катализаторами.
В седьмых, известно значительное количество ферментов, образующих с субстратами или промежуточными продуктами (в реакциях нуклеофильного замещения) временные ковалентные связи. В результате увеличивается скорость реакции от 100 до 1000 и более раз. В образовании временной ковалентной связи участвуют радикалы Гистидина, Серина, Цистеина (ковалентный катализ).
Рассмотренные элементы ферментативного катализа выявлены при изучении значительного количества различных ферментов, они обеспечивают чрезвычайную скорость реакций, показывают тонкость и сложность действия ферментов при минимальных затратах энергии в мягких условиях среды: температура до +40°С, рH около 7.
Для примера рассмотрим элементы механизма гидролиза -СО-NН-связи химотрипсином, который относится к сериновым протеазам. Химотрипсин гидролизует полипептидные цепи по карбонильной группе остатков аминокислот с гидрофобными, преимущественно ароматическими радикалами (Фенилаланин,Тирозин, Триптофан).
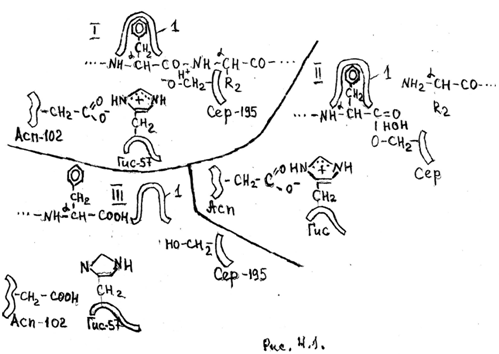 |
Каталитический центр химотрипсина состоит из трех главных аминокислотных остатков - Гис-57,Асп-102 и Сер-195. Схема их взаимного расположения и механизма действия представлены на рис. 4.1.
Схема активного центра сериновых протеаз и механизм их действия. 1-"якорная площадка" (полость) определяющая и закрепляющая радикалы аминокислотных остатков полипептидной цепи.
Протонирование имидозольного кольца Гис-57 осуществляется диссоциацией -СООН группы остатка Асп-102. Положительно заряженное имидозольное кольцо Гис-57 облегчает диссоциацию и ионизацию ОН- группы серина. Ионизация резко повышает нуклеофильный характер (донора электронной пары) остатка Сер -195 (I) который атакует углерод -СО- группы пептидной связи с отщеплением a-N -концевой половины полипептида и присоединением a-С-конца к радикалу Сер-195 в виде ацилфермента (II).На второй стадии ацилфермент гидролизуется с отщеплением a-С -концевой половины полипептида и регенерацией активного центра (III).
Поможем написать любую работу на аналогичную тему